We have now performed experiments in HT29 and HepG2 cells to test mitigation/synthetic lethality between…
04.30.20 progress update
While analysis of our CRISPR screens and initial RdRP experiments are still ongoing, we are making progress analyzing and following up on the RNA-seq data on remdesivir and HCQ host response.
HCQ: RNA-seq had identified upregulation of cholesterol metabolism genes as a consistent effect of HCQ treatment:
So we asked whether HCQ impairs cholesterol uptake due to its role as a endosome to lysosome trafficking inhibitor and found a strong HCQ-dependent reduction in LDL uptake:
We then asked whether this might lead to synthetic lethality with other drugs that impair cholesterol production. We identified robust synthetic lethality between HCQ and Simvastatin:
Next, we asked whether this HCQ impairment of cholesterol and this synthetic lethality with Simvastatin might be mitigated by cholesterol intermediates. We found that mevalonate, a cholesterol intermediate, could completely mitigate HCQ toxicity in HepG2 and HT29 cells, completely mitigate HCQ+Simvastatin lethality in HT29 and partially mitigate it in HepG2. CoQ10, a similar intermediate in cholesterol-related biosynthetic pathways, had similar efficacy in mitigation of HCQ toxicity but was less effective at mitigating combined HCQ+Simvastatin toxicity:
It is important to note that our findings at this point are on cancer cell lines, and so our current results that:
- HCQ cytotoxicity can be mitigated by mevalonate and CoQ10 (which has been tested clinically as a way to mitigate stain toxicity with mixed clinical trial results).
- HCQ+statins show synthetic lethality that can be partially alleviated with cholesterol intermediates.
must be taken with caution. Instead, such data should be combined with the CRISPR screening and human genetic association analyses we will perform. If gene-drug or drug-drug interactions are highlighted by these multiple orthogonal approaches, it raises confidence that they are robust and worth relaying to the medical community to assess their clinical value.
Remdesivir:
The two main pathways consistently impacted by remdesivir in the RNA-seq data are ER stress (ATF3 and ATF4-driven) and downregulation of mitochondrial respiration. ER stress seems to be non-specific (it is similarly affected by HCQ and likely just corresponds to generic effects of drugs on cells), so we have not pursued it further.
Regarding mitochondrial respiration, we see clear downregulation of all mitochondrial respiration complex genes:
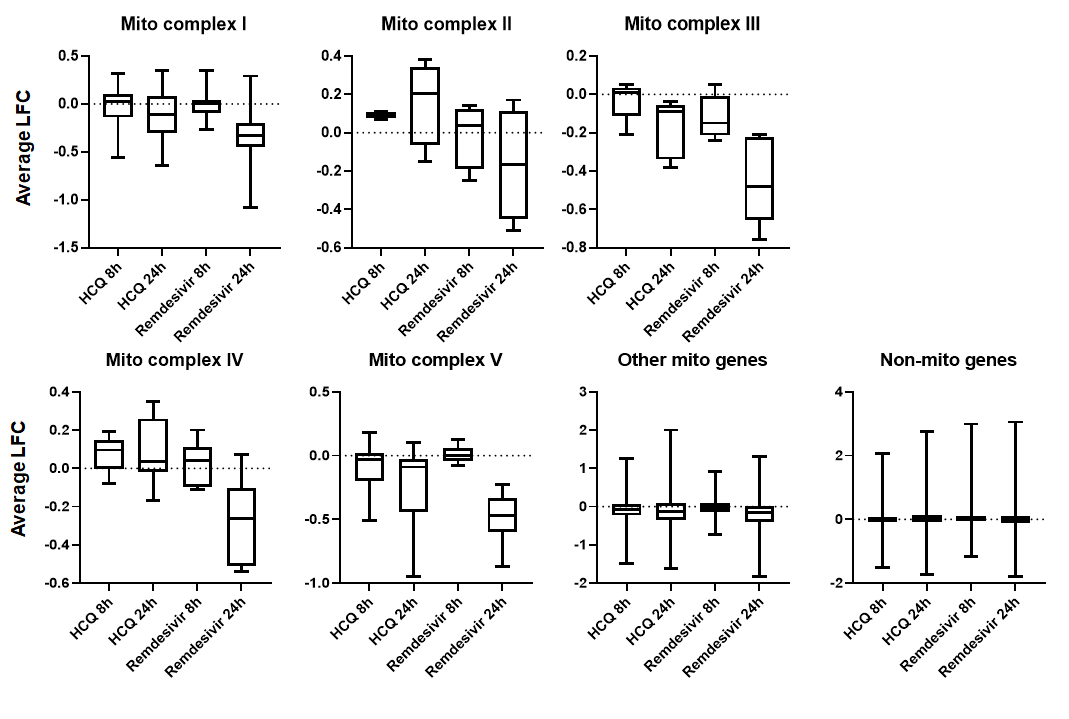
We asked whether other nucleoside analog drugs show similar effects. The only paper we could find on this topic (would love to hear of more) showed the opposite– the HIV drug AZT specifically upregulates mitochondrial respiration genes:
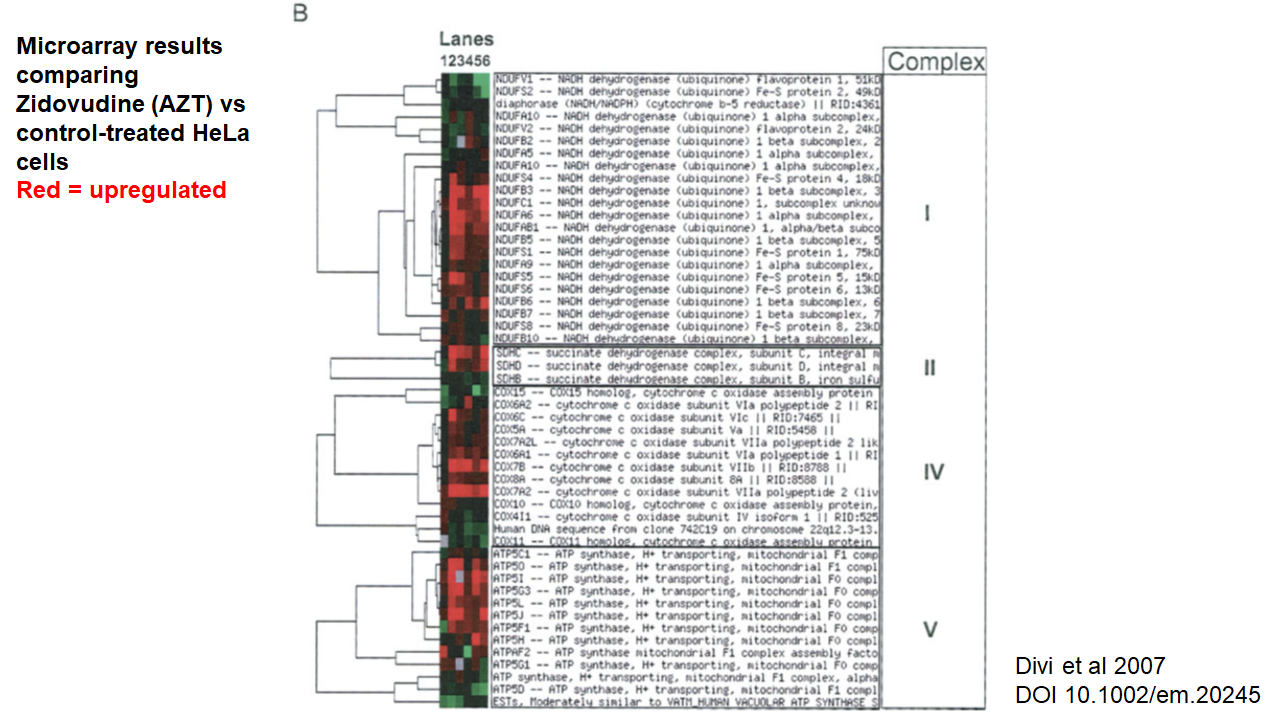
Nonetheless, we still believe that there is a good chance that the specific mechanism of remdesivir as an adenosine analog is likely responsible for this mitochondrial gene expression phenotype. Remdesivir is known to inhibit mitochondrial RNA polymerase, which may cause this effect.
Among the most downregulated genes are NFE2L3 (NRF3), a transcription factor whose other members NRF1 and NRF2 are well-known regulators of mitochondrial respiration, and UCP2, an oxidative stress responder that impacts mitochondrial respiration:
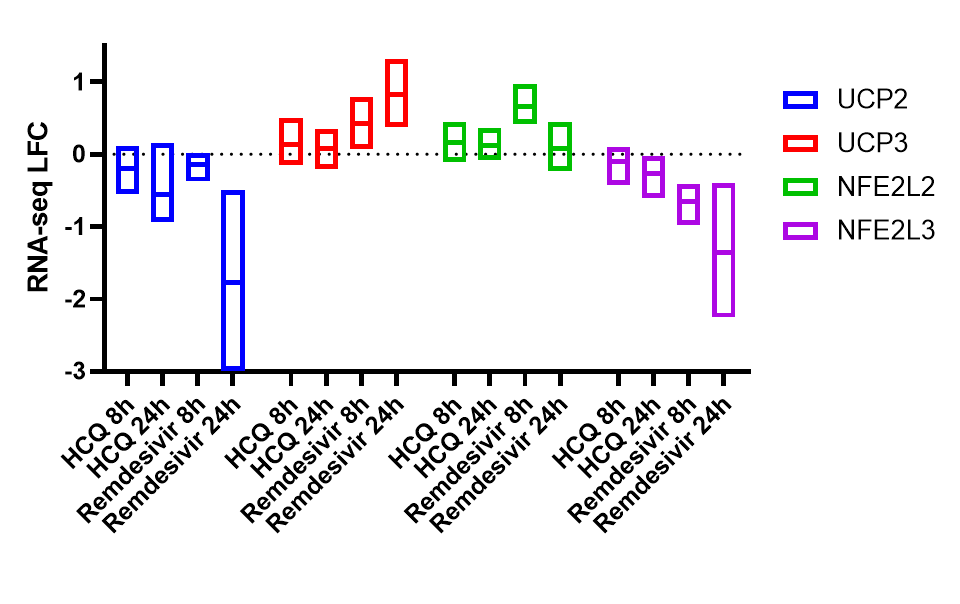
We are now pursuing whether modulators of mitochondrial respiration and specifically of NRF factors can mitigate remdesivir cytotoxicity (with the same caveats as above regarding cancer cell line data).